
Trained as an engineer, statistician, and computational biologist, I am passionate about providing principled solutions to analyzing complex data sets. My expertise can be broadly categorized into three domains:
I graduated with a Ph.D. in Electrical Engineering from Stanford in April 2020. I worked on statistics and machine learning with applications in biology and medicine, supervised by Professor Chiara Sabatti in the Departments of Statistics and Biomedical Data Science.
My projects have been highly interdisciplinary. I have also had multiple long-term collaborations with Professor Serafim Batzoglou in Computer Science and Professor Calvin Kuo in Medicine. I have been fortunate to apply my training in both engineering and statistics to new biotechnologies during my internships at 10x Genomics and Illumina.
I received my M.S. degree in Statistics from Stanford University and my B.S. degree in Electrical and Computer Engineering from Olin College of Engineering, where I worked with Professor Siddhartan Govindasamy, Professor Sarah Spence Adams, and Professor Denise Troxell. During my undergraduate studies, I was also a visiting research student with Professor Matthew McKay at Hong Kong University of Science and Technology.
I have had the good fortune to have many fruitful collaborations in academia and in industry. These projects would not have been possible without my extremely talented and hard-working collaborators.
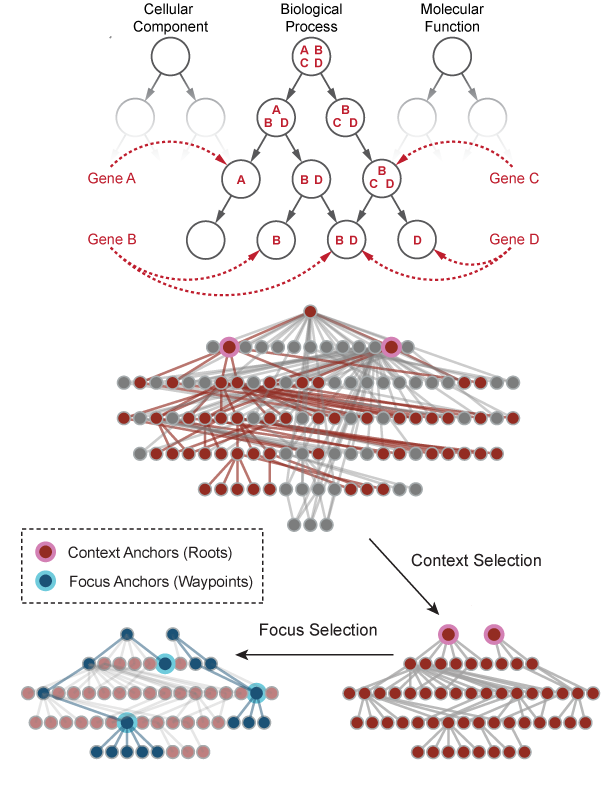
The Gene Ontology (GO) is one of the most popular biological knowledge databases for discovery-based omics research. Yet, the complexity of its data structure, represented as a large Directed Acyclic Graph (DAG) with tens of thousands of nodes, and its continuously evolving annotations can limit our interpretation and exploration in many applications.
I designed new algorithms and developed a full-stack software tool (AEGIS) to visualize the GO within an interactive framework. AEGIS overcomes the limitation of existing layout strategies that determine GO term specificity in the hierarchy by integrating both the GO structure and gene annotations into a new DAG layout. I also proposed an interactive visualization tailored for large DAGs by devising a focus-and-context framework, which can be used to navigate the DAG on a fine-grained level, while preserving contextual information of the entire DAG.
Website:
AEGIS and its visualization applications
Paper: Methodology behind AEGIS
Report: Project on D3 visualization
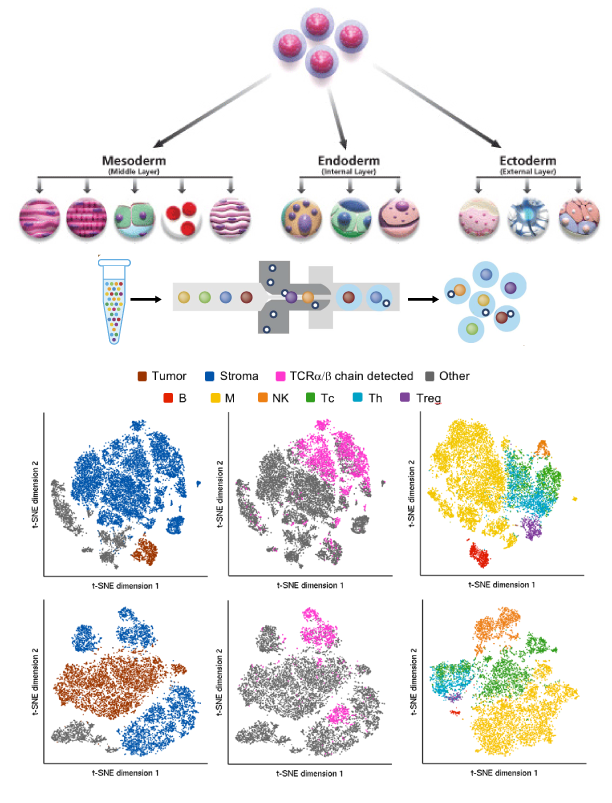
Single-cell technologies are a powerful means to measure gene expression levels of individual cells and to reveal previously unknown heterogeneity and functional diversity among cell populations. However, unknown biological variability and high measurement noise can make it difficult to analyze single-cell data.
Across multiple projects, I have developed new unsupervised learning (dimension reduction and clustering) techniques that are robust across sequencing platforms, analyzed over 50 data sets to build a comprehensive database, and contributed to a number of computational and statistical pipelines for new biotechnologies and applications. A lot of our techniques also extend to other biological data analysis. I have worked on single-cell applications that span immunology, oncology, and stem cell biology.
Paper 1: The tumor immune microenvironment
Paper 2: Long-term expansion of primary hepatocytes
Paper 3: SIMLR: kernel-based similarity learning
Paper 4:
Single-cell RNA-seq database
Paper 5: 10x Genomics droplet technology
Paper 6: Extended application to Hi-C data
Code 1: R, Python and MATLAB code for SIMLR
Code 2: R scripts for 10x droplet technology
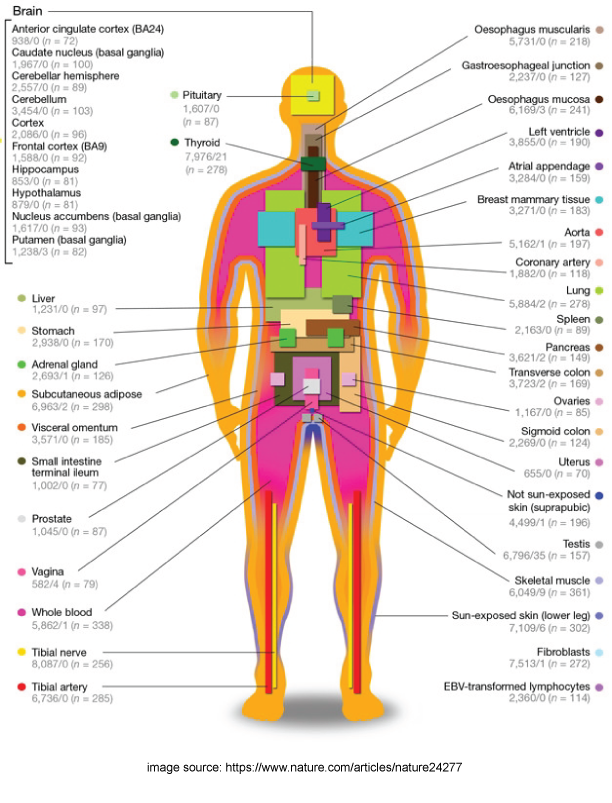
The Genotype-Tissue Expression (GTEx) project aims to discover associations between individual genetic variance and the gene expression of different tissues. It consists of 53 types of human tissues from healthy, postmortem donors who were densely genotyped. These tissues are costly to acquire, so one of the main challenges is detecting signals with a very limited sample size.
I developed systematic analysis pipelines to conduct expression quantitative trait loci (eQTLs) studies where the goal is to identify tissue-specific gene expression that is highly correlated with genetic mutation.s With my collaborators, we proposed a selective inference analysis workflow to account for selective biases when analyzing a large number of genes, which can provide accurate confidence interval coverages with limited sample sizes. Additionally, I worked on gene function prediction problems with the same dataset as part of a data mining course project.
Paper: Selection-adjusted Effect-size Estimation
Report: Project report on gene function prediction
I am grateful to my undergraduate mentors who patiently taught me everything from scratch and got me excited about research early on. This theoretical foundation built my intuition for many applied projects in other fields during my graduate studies.
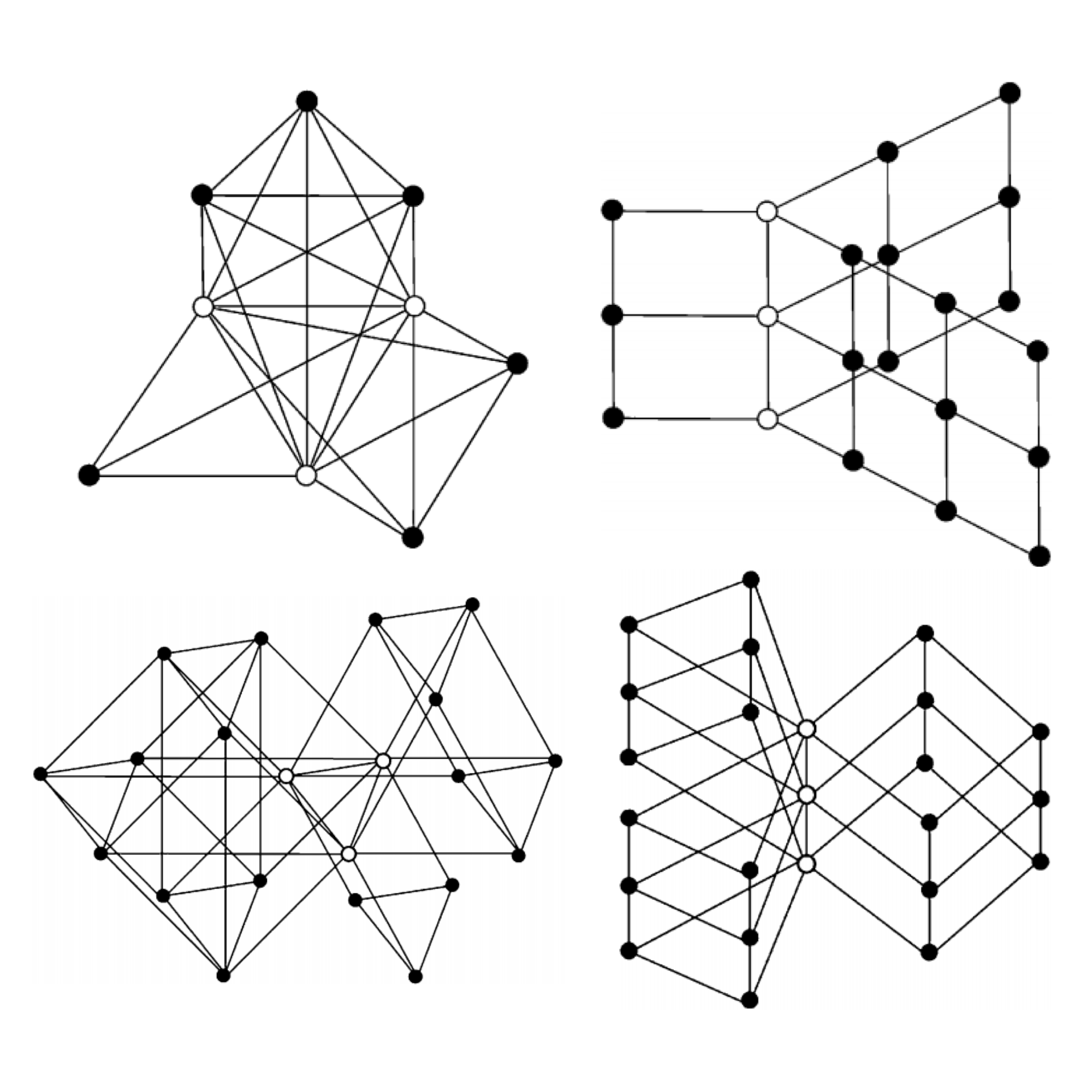
The L(2,1)-labeling of a graph is an extension of the classical graph coloring problem. It is motivated by assigning frequencies to transmitters in a radio network. By investigating a wide family of graphs and enumerating many example graphs, we found general labeling patterns and unique graph structures. In addition, we proved a conjecture in a related coloring problem.
Paper 1: Amalgamation Graphs
Paper 2: Edge-path Replacement Graphs
Paper 3: Injective Labeling of General Graphs
Paper 4: Cartesian Products of Complete Graphs
Paper 5: Generalized Flowers
Using stochastic geometry for the theoretical analysis of wireless networks, we statistically characterized the performance of multi-antenna receivers under more general network topologies than what was available at the time. In addition, analyzing the impact of carrier frequency offsets on multi-user systems that apply orthogonal frequency-division multiplexing, I derived analytical results that determine the impact of unsynchronized interferers.
Paper 1: Non-homogenous Poisson Networks
Paper 2: Networks with Clustered Interferers
Paper 3: Multi-antenna OFDM Systems
Paper 4: Doubly Stochastic Networks
Only peer-reviewed papers are listed here. An extended list can be found at my Google Scholar page.
J. Zhu, Q. Zhao, E. Katsevich, C. Sabatti, “Exploratory gene ontology analysis with interactive visualization,” Scientific Reports, Vol 1, No. 1, pp.7793, 2019.
S. Panigrahi, J. Zhu, C. Sabatti, “Selection-adjusted inference: an application to confidence intervals for cis-eQTL effect sizes,” Biostatistics, kxz024.
J. T. Neal†, X. Li†, J. Zhu, ..., C. Sabatti, J. S. Boehm, W. C. Hahn, G. X.Y. Zheng, M. M. Davis, C. J. Kuo, “Organoid modeling of the tumor immune microenvironment,” Cell, Vol. 175, No. 7, pp. 1972-1988, 2018.
W. C. Peng, C. Y. Logan, M. Fish, T. Anbarchian, F. Aguisanda, A. Alvarez-Varela, P. Wu, Y. Jin, J. Zhu, B. Li, M. Grompe, B. Wang, R. Nusse, “Inflammatory cytokine TNFα promotes the long-term expansion of primary hepatocytes in 3D culture,” Cell, Vol. 175, No. 6, pp. 1607-1619, 2018.
B. Wang†, A. Pourshafeie†, M. Zitnik†, J Zhu, C. D. Bustamante, J. Leskovec, S. Batzoglou, “Network Enhancement: a general method to denoise weighted biological networks,” Nature Communications, Vol. 9, No. 1, pp. 3108, 2018.
B. Wang, D. Ramazzotti, L. De Sano, J. Zhu, E. Pierson, S. Batzoglou, “SIMLR: A Tool for Large-Scale Genomic Analyses by Multi-Kernel Learning,” Proteomics, Vol. 18, No. 2, pp. 1700232, 2018.
Y. Cao†, J. Zhu†, G. Han, P. Jia, Z. Zhao “scRNASeqDB: a database for gene expression profiling in human single-cell by RNA-seq,” Genes, Vol. 8, No. 12, pp. 368, 2017.
B. Wang, J. Zhu, E. Pierson, D. Ramazzotti, S. Batzoglou, “Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning,” Nature Methods, Vol. 14, No. 4, pp. 414, 2017.
G. X.Y. Zheng, J. M Terry, P. Belgrader, P. Ryvkin, Z. W. Bent, R. Wilson, S. B. Ziraldo, T. D. Wheeler, G. P. McDermott, J. Zhu, ..., T. S. Mikkelsen, B. J. Hindson, J. H. Bielas, “Massively parallel digital transcriptional profiling of single-cells,” Nature Communications, Vol. 10, No. 11, pp. 1096-1098, 2017.
B. Wang, J. Zhu, Oana Ursu, Armin Pourshafeie, Serafim Batzoglou, Anshul Kundaje, “Unsupervised learning from noisy networks with applications to Hi-C data',” in Proceedings of Advances in Neural Information Processing Systems (NIPS), Barcelona, Dec. 2016.
J. Zhu, S. Govindasamy, J. Hwang, “Performance of multiantenna linear MMSE receivers in doubly stochastic networks,” IEEE Transactions on Communications, Vol. 62, No.8, pp.2825,2839, Aug. 2014.
J. Zhu, R. H. Y. Louie, M. R. McKay, S. Govindasamy, “On the impact of unsynchronized interferers on multi-antenna OFDM systems,” in Proceedings of the IEEE International Conference on Communications (ICC), Sydney, June 2014.
J. Zhu, S. Govindasamy, “Performance of multi-antenna linear MMSE receivers in the presence of clustered interferers,” in Proceedings of the IEEE International Conference on Communications (ICC), Budapest, June 2013.
J. Zhu, S. Govindasamy, “Performance of multi-antenna MMSE receivers in non-homogenous Poisson networks,” in Proceedings of the IEEE International Conference on Communications (ICC), Ottawa, June 2012.
N. Karst, J. Oehrlein, D. S. Troxell, J. Zhu, “The minimum span of L(2,1)-labelings of generalized flowers,” Discrete Applied Mathematics, Vol. 181, pp. 139-151, January 2015.
N. Karst, J. Oehrlein, D. S. Troxell, J. Zhu, “Labeling amalgamations of Cartesian products of complete graphs with a condition at distance two,” Discrete Applied Mathematics, Vol. 178, pp. 101-108, December, 2014.
N. Karst, J. Oehrlein, D. S. Troxell, J. Zhu, “On distance labelings of amalgamations and injective labelings of general graphs,” Involve, a Journal of Mathematics, Vol. 8, No. 4, pp. 535-540, 2014.
N. Karst, J. Oehrlein, D. S. Troxell, J. Zhu, “L(d, 1)-labelings of the edge-path-replacement by factorization of graphs.” Journal of Combinatorial Optimization, May 2013.
S. S. Adams, N. Howell, N. Karst, D. S. Troxell and J. Zhu, “On the L(2, 1)-labelings of amalgamations of graphs,” Discrete Applied Mathematics, Vol. 161, No. 7–8, pp.881-888, May 2013