Jason Zhu, Ph.D.
Bridging research depth and product impact
Bridging research depth and product impact
I am a curiosity-driven, scientifically trained builder with 10+ years of experience in AI/ML, statistics, and graph algorithms. I have had the fortune to collaborate with world-class researchers and top-tier product teams to drive meaningful, collective impact—reflected in 10,000+ citations to my publications. Passionate about complex challenges and high-agency environments, I architect and implement scalable solutions across emerging domains, from multi-modal RAGs and intelligent search to biomedical discovery.
I earned my Ph.D. in Electrical Engineering from Stanford University, where I discovered how statistics and ML can help us understand complex systems in biology and health. I also collaborated with researchers and startups on projects ranging from DNA analysis to interactive data visualizations.
After Stanford, I joined Apple and worked on improving how apps like Maps and Search adapt to user preferences. I built tools to automatically test and fix issues in ML systems, making results smarter and user experiences better. I also briefly worked in the startup world at Nexa AI, leading efforts to build private, on-device AI systems.
A decade of building tools, uncovering structure, and chasing ideas across biology, engineering, and machine learning.
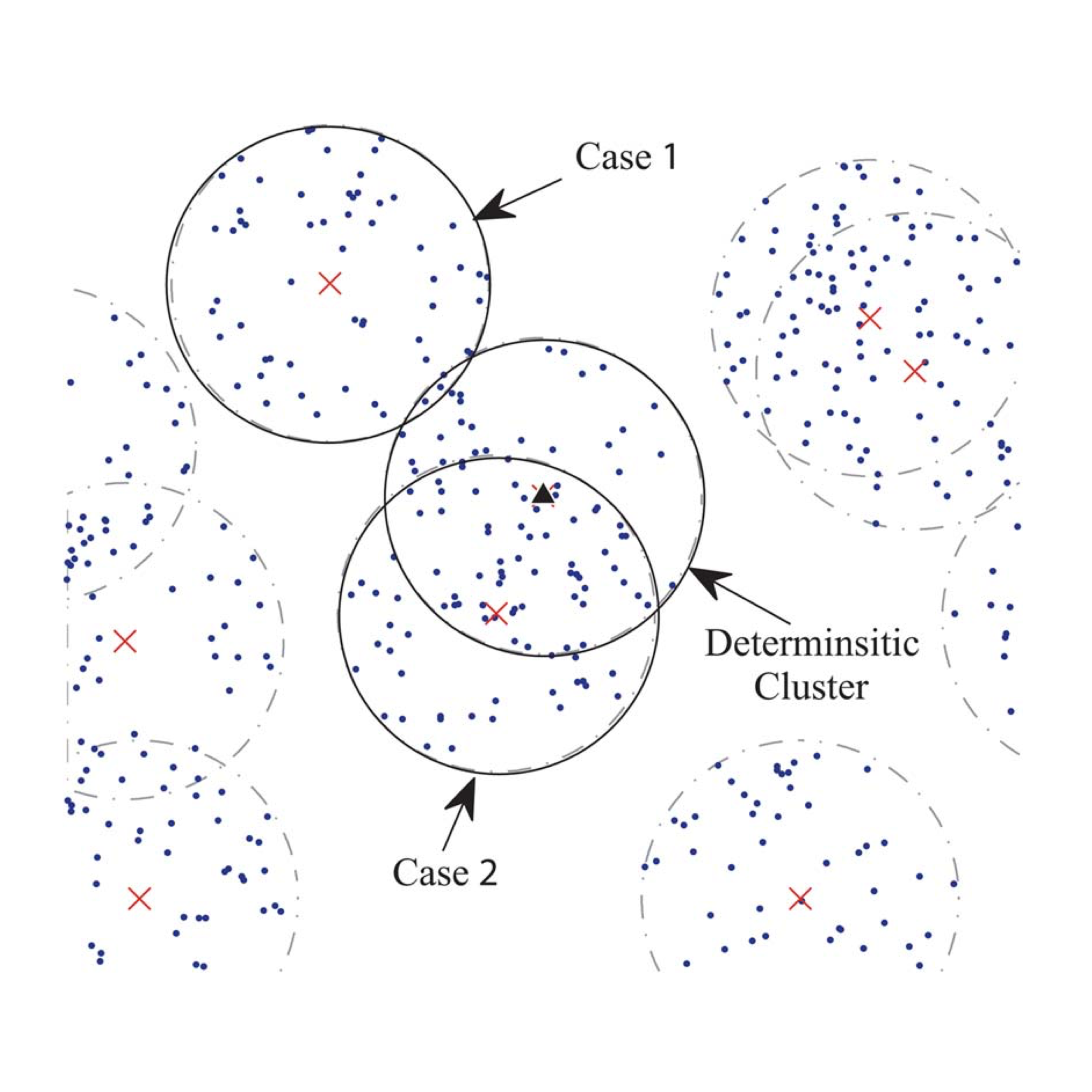
The Gene Ontology (GO) is one of the most popular biological knowledge databases for discovery-based omics research. Yet, the complexity of its data structure, represented as a large Directed Acyclic Graph (DAG) with tens of thousands of nodes, and its continuously evolving annotations can limit our interpretation and exploration in many applications.
I designed new algorithms and developed a full-stack software tool (AEGIS) to visualize the GO within an interactive framework. AEGIS overcomes the limitation of existing layout strategies that determine GO term specificity in the hierarchy by integrating both the GO structure and gene annotations into a new DAG layout. I also proposed an interactive visualization tailored for large DAGs by devising a focus-and-context framework, which can be used to navigate the DAG on a fine-grained level, while preserving contextual information of the entire DAG.
Website:
AEGIS and its visualization applications
Paper: Methodology behind AEGIS
Report: Project on D3 visualization
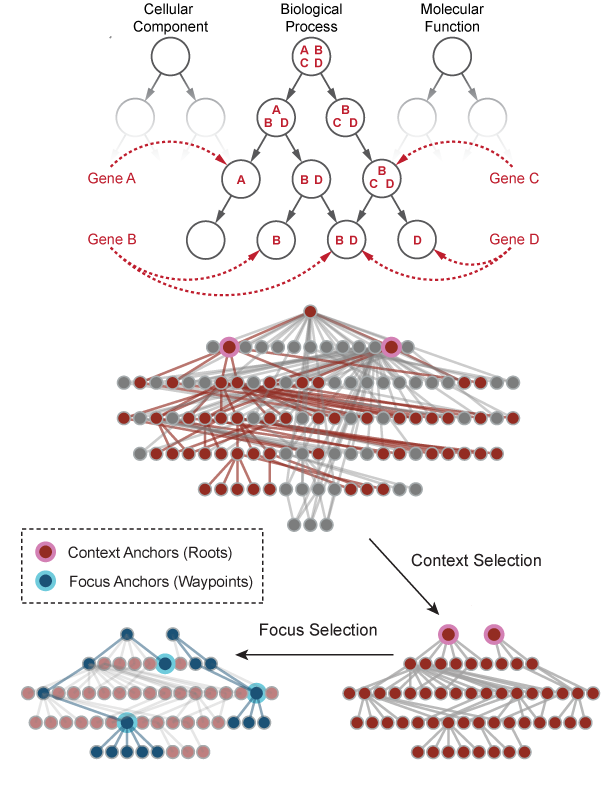
Single-cell technologies are a powerful means to measure gene expression levels of individual cells and to reveal previously unknown heterogeneity and functional diversity among cell populations. However, unknown biological variability and high measurement noise can make it difficult to analyze single-cell data.
Across multiple projects, I have developed new unsupervised learning (dimension reduction and clustering) techniques that are robust across sequencing platforms, analyzed over 50 data sets to build a comprehensive database, and contributed to a number of computational and statistical pipelines for new biotechnologies and applications. A lot of our techniques also extend to other biological data analysis. I have worked on single-cell applications that span immunology, oncology, and stem cell biology.
Paper 1: The tumor immune microenvironment
Paper 2: Long-term expansion of primary hepatocytes
Paper 3: SIMLR: kernel-based similarity learning
Paper 4:
Single-cell RNA-seq database
Paper 5: 10x Genomics droplet technology
Paper 6: Extended application to Hi-C data
Code 1: R, Python and MATLAB code for SIMLR
Code 2: R scripts for 10x droplet technology
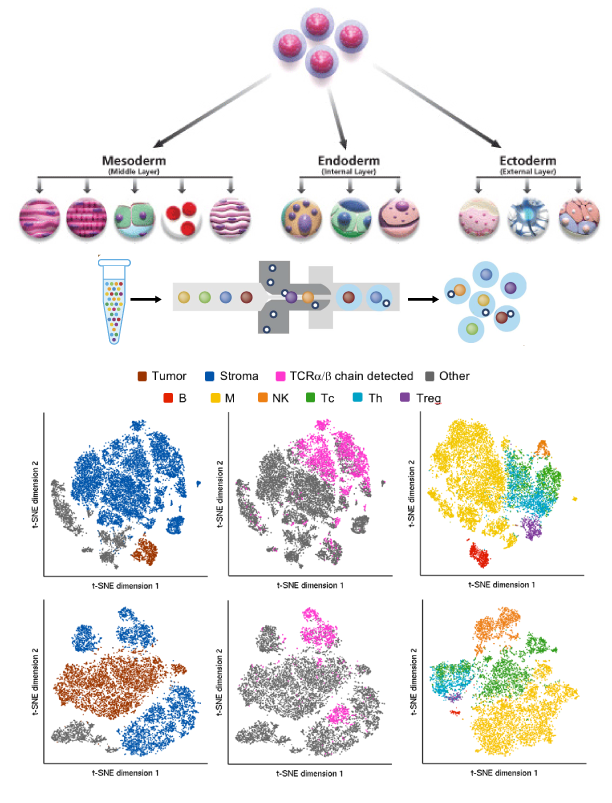
The Genotype-Tissue Expression (GTEx) project aims to discover associations between individual genetic variance and the gene expression of different tissues. It consists of 53 types of human tissues from healthy, postmortem donors who were densely genotyped. These tissues are costly to acquire, so one of the main challenges is detecting signals with a very limited sample size.
I developed systematic analysis pipelines to conduct expression quantitative trait loci (eQTLs) studies where the goal is to identify tissue-specific gene expression that is highly correlated with genetic mutation.s With my collaborators, we proposed a selective inference analysis workflow to account for selective biases when analyzing a large number of genes, which can provide accurate confidence interval coverages with limited sample sizes. Additionally, I worked on gene function prediction problems with the same dataset as part of a data mining course project.
Paper: Selection-adjusted Effect-size Estimation
Report: Project report on gene function prediction
The L(2,1)-labeling of a graph is an extension of the classical graph coloring problem. It is motivated by assigning frequencies to transmitters in a radio network. By investigating a wide family of graphs and enumerating many example graphs, we found general labeling patterns and unique graph structures. In addition, we proved a conjecture in a related coloring problem.
Paper 1: Amalgamation Graphs
Paper 2: Edge-path Replacement Graphs
Paper 3: Injective Labeling of General Graphs
Paper 4: Cartesian Products of Complete Graphs
Paper 5: Generalized Flowers
Using stochastic geometry for the theoretical analysis of wireless networks, we statistically characterized the performance of multi-antenna receivers under more general network topologies than what was available at the time. In addition, analyzing the impact of carrier frequency offsets on multi-user systems that apply orthogonal frequency-division multiplexing, I derived analytical results that determine the impact of unsynchronized interferers.
Paper 1: Non-homogenous Poisson Networks
Paper 2: Networks with Clustered Interferers
Paper 3: Multi-antenna OFDM Systems
Paper 4: Doubly Stochastic Networks